Revising RNA Force Fields for Molecular Mechanics Calculations
Theories for important interactions are required to rationalize experimental results and predict the secondary and three dimensional structures of RNA molecules. Quantum mechanics (QM) is the best theory for describing the interactions in atomic systems but even the simplest RNA systems have hundreds of atoms that do not allow the direct application of ab initio calculations. Even though QM cannot be applied directly to RNAs it can be applied to individual components such as guanine, adenine, uracil, and cytosine, and those results can be used to build models to predict experimental observables. These models are called “force fields”. Most of the popular force fields include an atom-centered point-charge model. These simulation methods need a set of point charges for all the atoms in the system. Starting with an initial structure and using a force field, properties such as stability and dynamics can be predicted for complicated RNA configurations.
Understanding the physical interactions governing the structure and dynamics of ribonucleotides should improve the accuracy of simulations of ribonucleic acid (RNA) molecules. Methods for simulating biological systems include residue centered force fields (coarse-grained), atom-centered force fields (AMBER, CHARMM, GROMOS), approximate quantum mechanics, and mixed quantum mechanics/molecular mechanics methods (QM/MM). With advances in computer power, it is possible to run simulations at least as long as milliseconds and microseconds with coarse-grained and atom-centered potentials, respectively. The AMBER force fields are particularly widely used for simulations of RNA. They have provided satisfactory descriptions of structural and thermodynamic properties for some RNA and deoxyribonucleic acid (DNA) systems, while some challenging systems still provide difficulty. We published a review article back in 2005 in Biochemistry entitled “RNA challenges for computational chemists” where we discussed some of the important experimental results needing theoretical explanations. At that time, the RNA force fields could simulate regular RNA duplexes while properties of exotic RNA systems were a challenge to predict using computational methods. Thus, RNA force fields require revisions in order to reconcile predictions with experimental measurements.
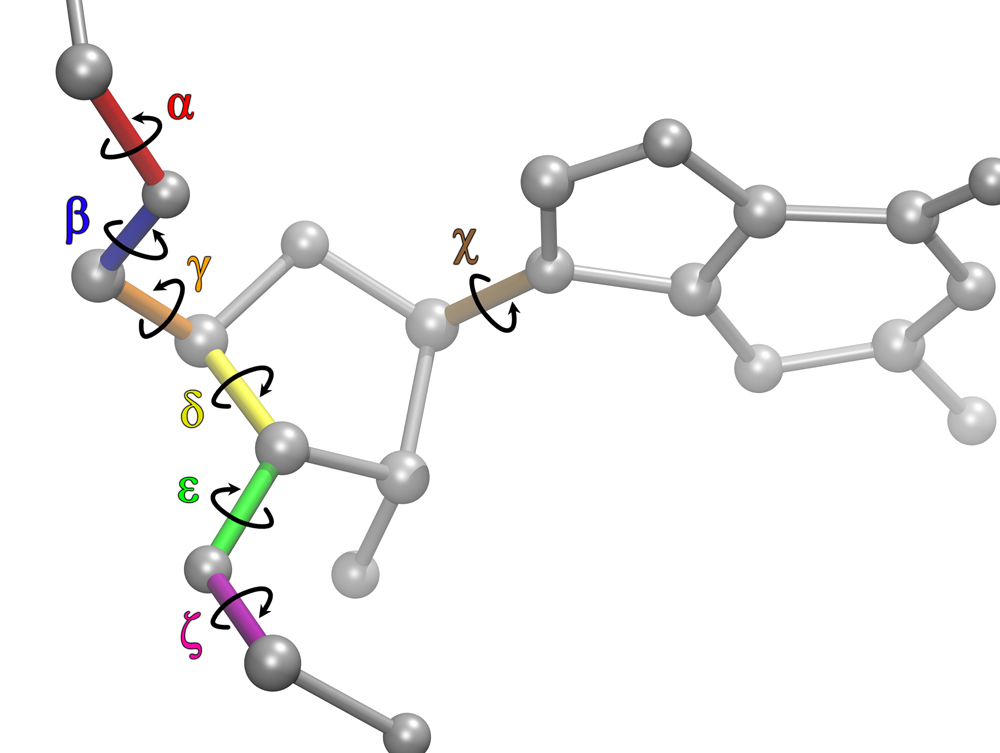 In our initial work we discovered that RNA force fields required revisions in order explain the experimental results observed in single-stranded RNA and RNA with
modified residues. The main reason for this big discrepancy was due to the bad parameterization of the χ torsional parameters. There were several reasons why amber99
could not predict the experimental results for these systems: 1) When the χ torsions were parametrized for amber99, model systems for adenosine and thymidine were used,
and the results were generalized for all DNA and RNA residues. 2) Model systems they used in the parameterization process mimicked deoxyribose C2′-endo sugar puckering.
3) At that time, QM calculations were limited by computer power and only 8-9 data points were used in the QM fitting. 4) In the amber99 force field, the original Cornell
force field parameters for χ torsions were changed without doing any fitting to improve the C2′-endo sugar puckering phase angle for DNA residues. All these combined
will change the potential energy surface of the RNA nucleosides, which would not represent the correct energy landscape of RNA molecules.
In our initial work we discovered that RNA force fields required revisions in order explain the experimental results observed in single-stranded RNA and RNA with
modified residues. The main reason for this big discrepancy was due to the bad parameterization of the χ torsional parameters. There were several reasons why amber99
could not predict the experimental results for these systems: 1) When the χ torsions were parametrized for amber99, model systems for adenosine and thymidine were used,
and the results were generalized for all DNA and RNA residues. 2) Model systems they used in the parameterization process mimicked deoxyribose C2′-endo sugar puckering.
3) At that time, QM calculations were limited by computer power and only 8-9 data points were used in the QM fitting. 4) In the amber99 force field, the original Cornell
force field parameters for χ torsions were changed without doing any fitting to improve the C2′-endo sugar puckering phase angle for DNA residues. All these combined
will change the potential energy surface of the RNA nucleosides, which would not represent the correct energy landscape of RNA molecules.
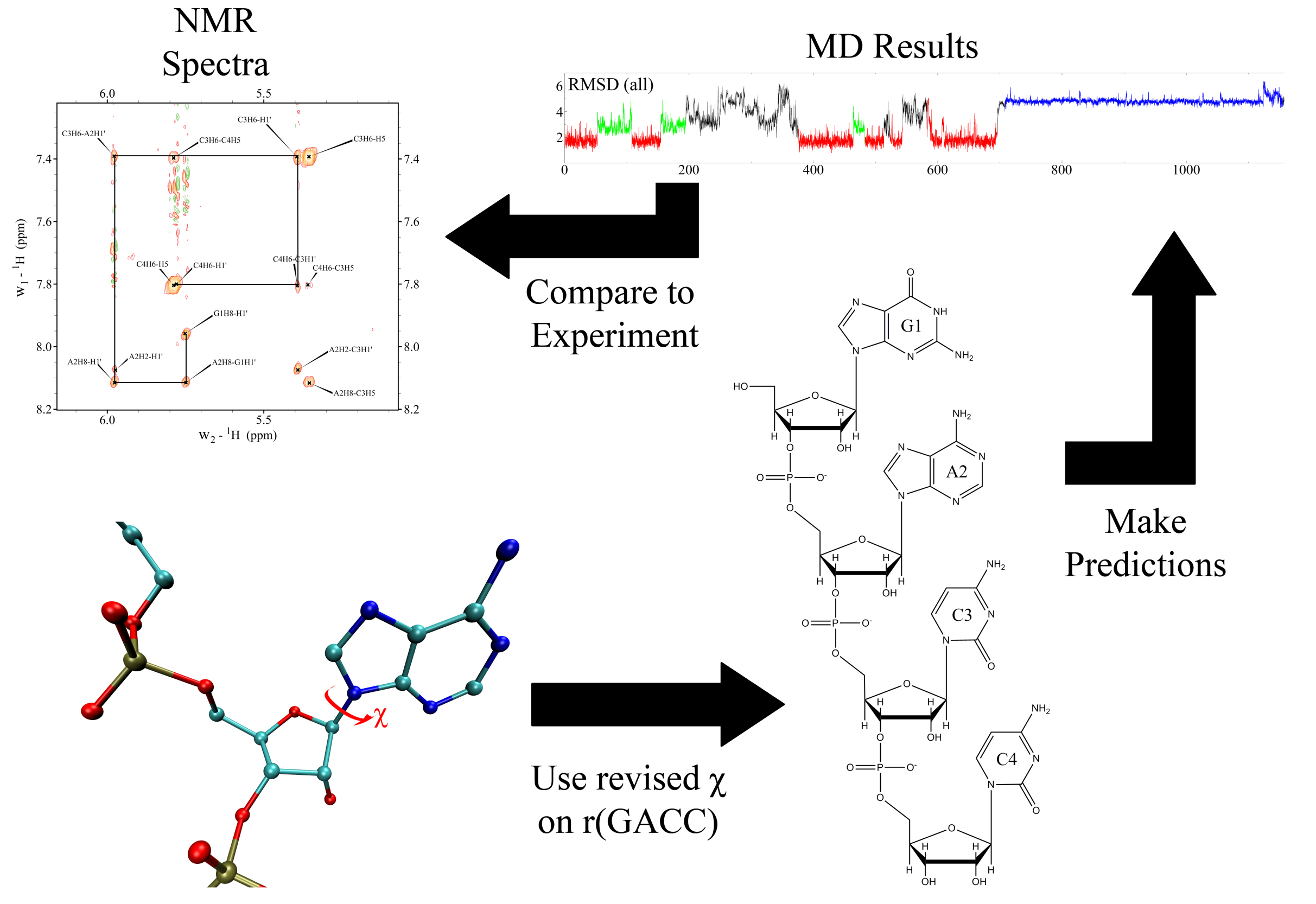
Thus, to solve the problems with RNA force fields, we revised the χ, β, ε, and ζ torsional parameters for AMBER force field, and did NMR spectroscopy on RNA mononucleosides, single-stranded RNA GACC, and an RNA duplex with modified bases, (iGiGiCiC)2. Moreover, we developed a new mixing rule for Thermodynamic Integration Approach, a method to make free energy predictions, and implemented it to AMBER version 9. Results showed that the revised RNA force field revisions dramatically improved the predictions. The revised parameters increased agreement with experiment by 10,000,000 – fold relative to results with the commonly used parameters. Furthermore, our work stimulated other researchers to revise the torsional parameters of RNA and DNA. We further revised the force field parameters for modified RNA residues, such as locked nucleic acids (LNA) and 2’-O-methyl RNA residues, which are widely used in cancer research. Recently we showed also that the α/γ torsional parameters for RNA require revisions to resolve issues seen in MD simulations of single-stranded RNA molecules. Altogether, our long term goal is to create models for RNA so that predictions are physical, and can be used to understand molecular properties of RNA systems.
© YildirimLab 2017